Home
News
Research
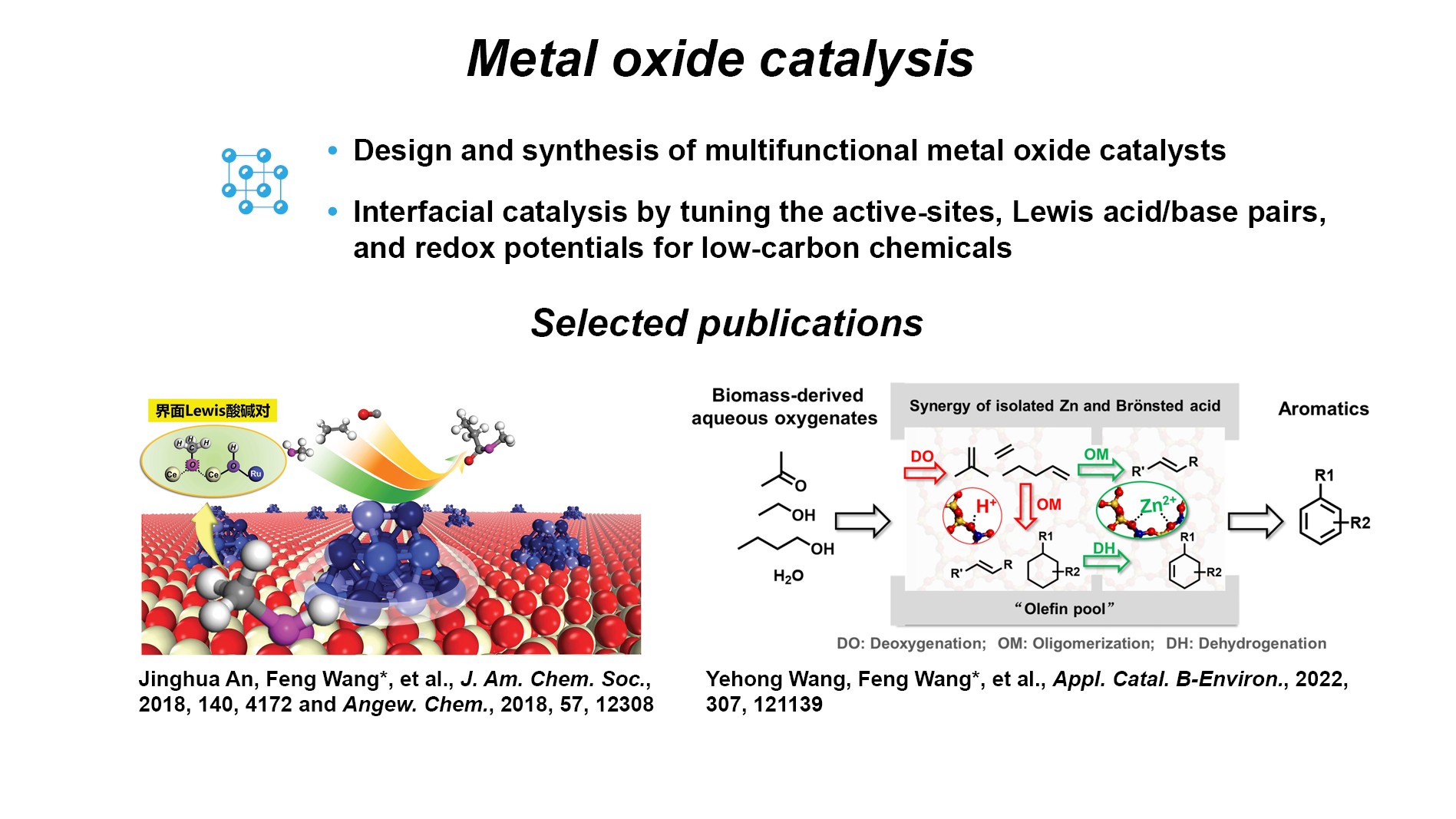
Metal oxide catalysis
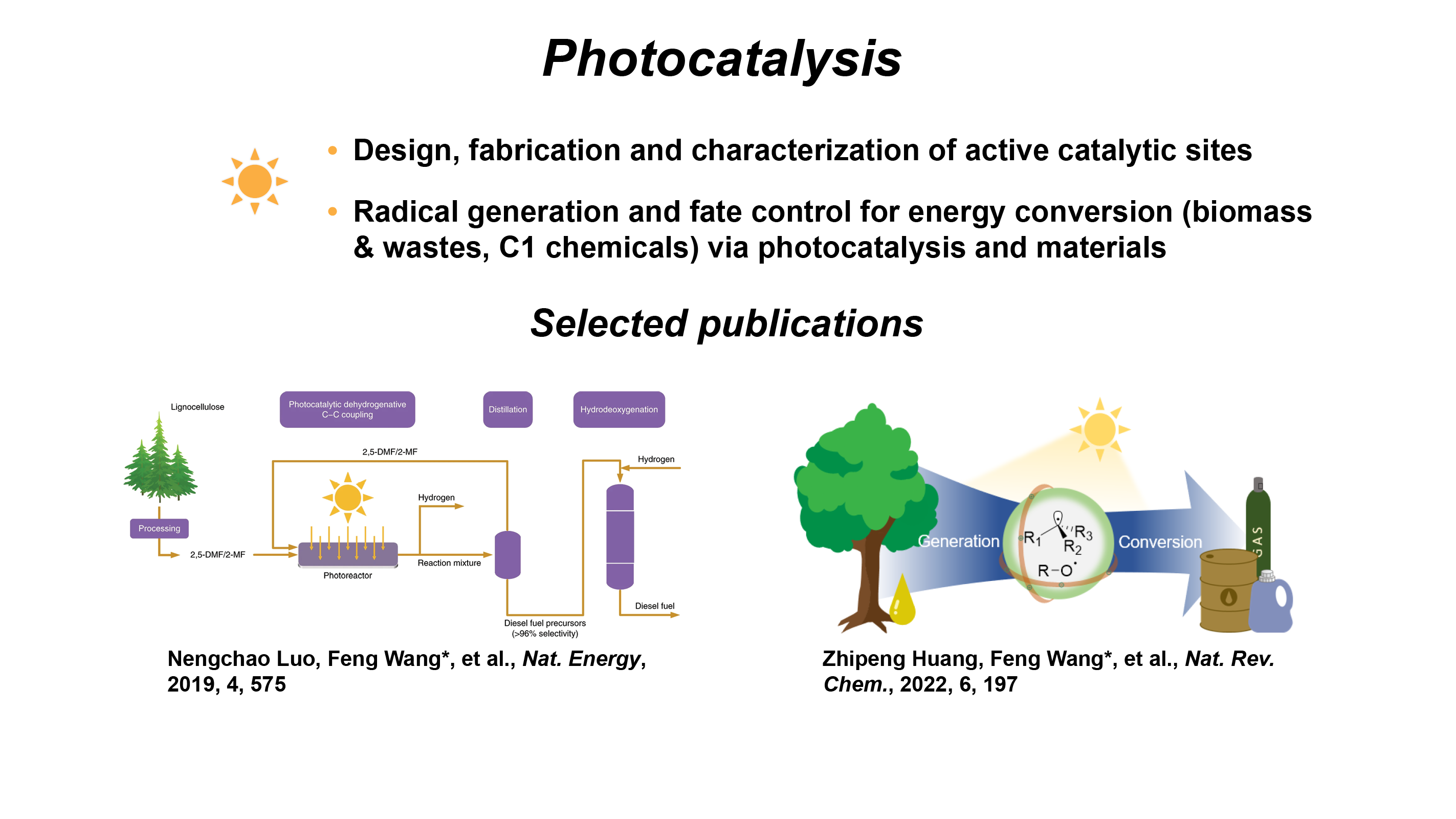
Photocatalysis
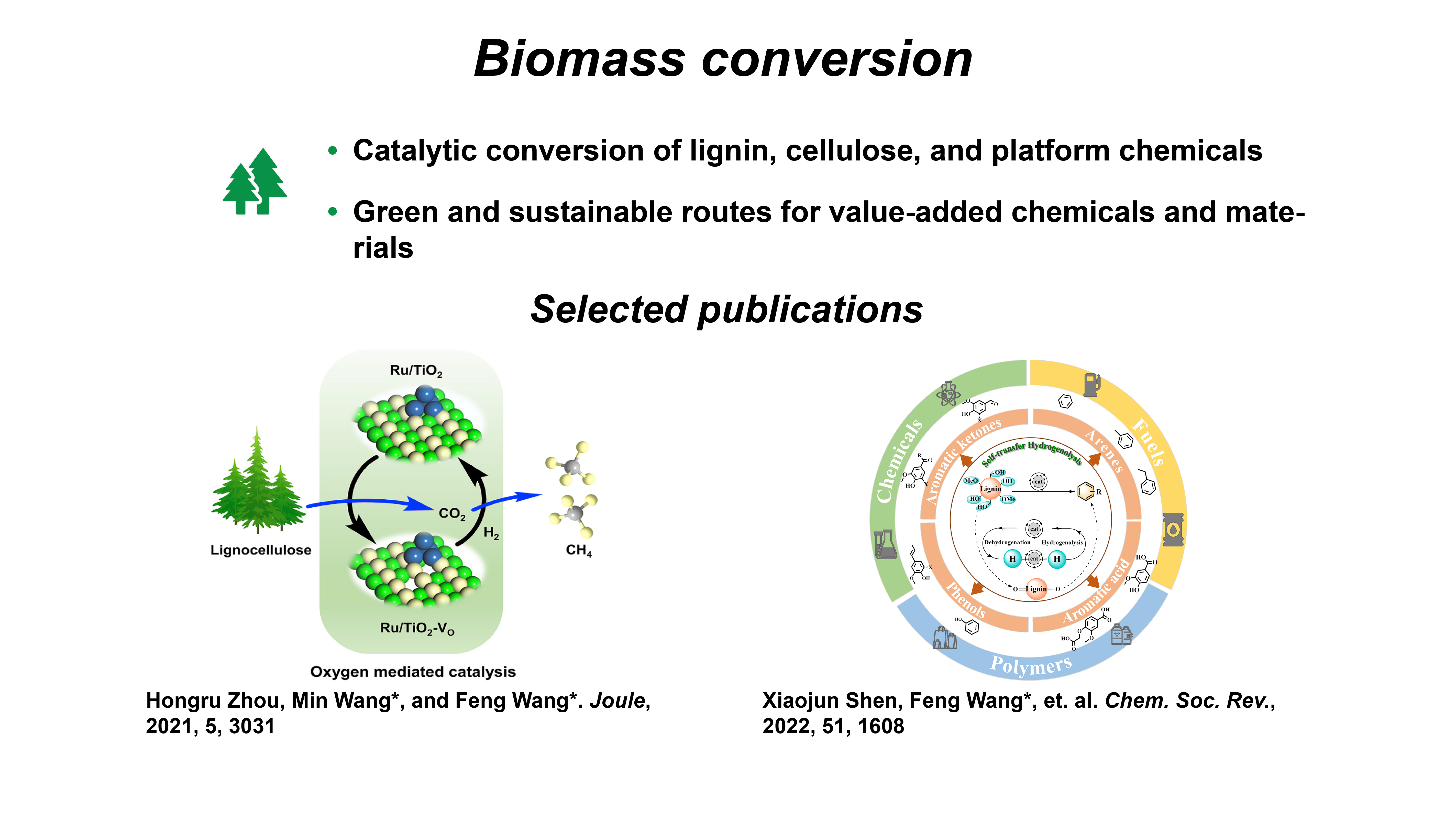
Biomass conversion
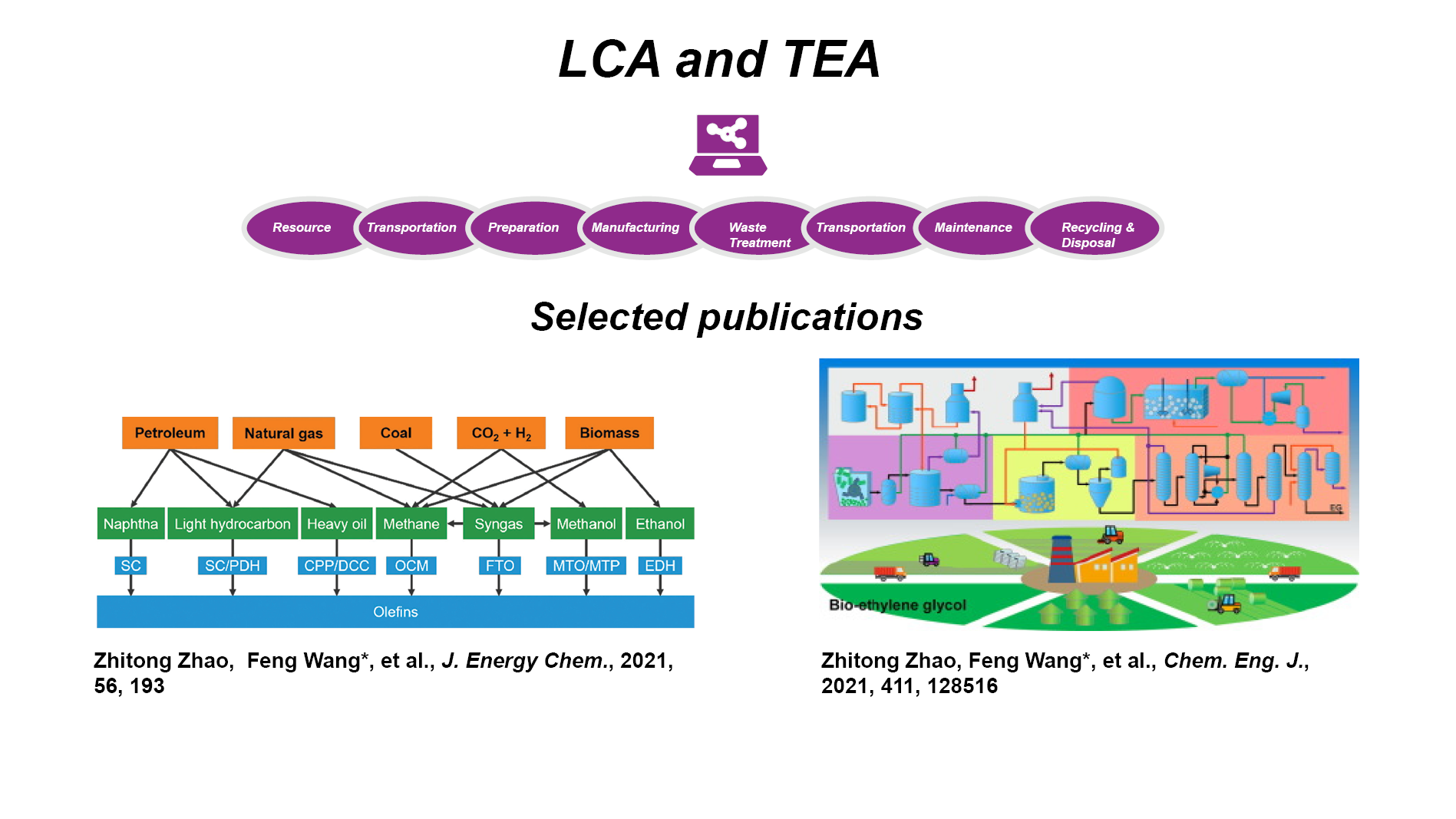
LCA & TEA
Team
Director
Staffs
Postdocs
Graduate Students
Alumni
Publications
2025
2024
2023
2022
2021
2020
2019
2018
2017
2016
A full list
Visuals
Group Photos
Videos
Contact
Link
MENU
Research
Metal oxide catalysis
Photocatalysis
Biomass conversion
LCA & TEA